This Case of the Month details our comprehensive management of a 29-year-old male with the rare X-linked tubulopathy, Dent disease (DD).
The patient presented with two obstructing right ureteral stones along with significant bilateral nephrolithiasis and nephrocalcinosis—totaling over 5 cm of stone burden in each kidney. Using a staged surgical approach combining ureteroscopy (URS) and percutaneous nephrolithotomy (PCNL) with advanced suction sheath technology, we achieved complete stone clearance within one month while preserving renal parenchyma and maximizing kidney function.
While surgical management of kidney stones in DD follows standard principles used for complex nephrolithiasis, there are currently no formal guidelines for the medical management of DD. Therefore, we also provide a detailed overview of our diagnostic approach to DD and therapeutic considerations.
We hope this report serves as a practical resource for clinicians managing similar complex cases.
Case Highlights:
- The patient had stage 4 CKD with a GFR of 17.7 mL/min/1.73 m², proteinuria, and significant bilateral nephrolithiasis/nephrocalcinosis—all keystone findings of DD.
- Management involved staged bilateral PCNLs, with initial right combo URS/PCNL followed by left PCNL three weeks later.
- Advanced vacuum-assisted nephrostomy sheath technology minimized intrarenal pressure and maximized stone-free rate.
- The case highlights clinical characteristics, diagnostic testing, and medical management of DD and provides context to this rare genetic disease.
Background on Dent Disease
DD is a rare X-linked recessive condition characterized by significant proteinuria, hypercalciuria, nephrolithiasis, nephrocalcinosis, and kidney failure usually in the third to fifth decades of life in men. First described by Drs. Charles Dent and Max Friedman in 1964, the genetic disease is caused by mutations in the CLCN5 gene (60 percent of patients) and OCRL1 gene (15 percent of patients), both of which encode a kidney-specific chloride channel1,2.
DD can be difficult to diagnose due to lack of obvious symptoms. Proteinuria is identified as nephrotic syndrome while rickets or nephrolithiasis are diagnosed in other disease process. A combination of hypercalciuria, nephrocalcinosis, nephrolithiasis, proteinuria without nephrotic syndrome (normal serum albumin), and unexplained chronic kidney disease (CKD) in young men are symptoms that should prompt investigation for DD, especially with family history of kidney failure in maternal male relatives3. Definitive diagnosis is confirmed by genetic testing.
Patient Case
The patient was diagnosed with DD at age 17 and was referred for definitive management of substantial urolithiasis. He first passed stones at age 15 and had URS with laser lithotripsy at age 23 and again at 24. His family history is significant for a younger brother with DD and nephrolithiasis. His maternal grandfather died of kidney failure in his early 50s and likely had DD. He is actively managed by a nephrologist on thiazide diuretics, angiotensin converting enzyme (ACE) inhibition, and citrate supplementation for several years.
At presentation, his stone burden had increased from imaging collected two years prior and he reported experiencing intermittent renal colic, though was asymptomatic at time of the initial office visit. A review of systems was negative at time of consultation.
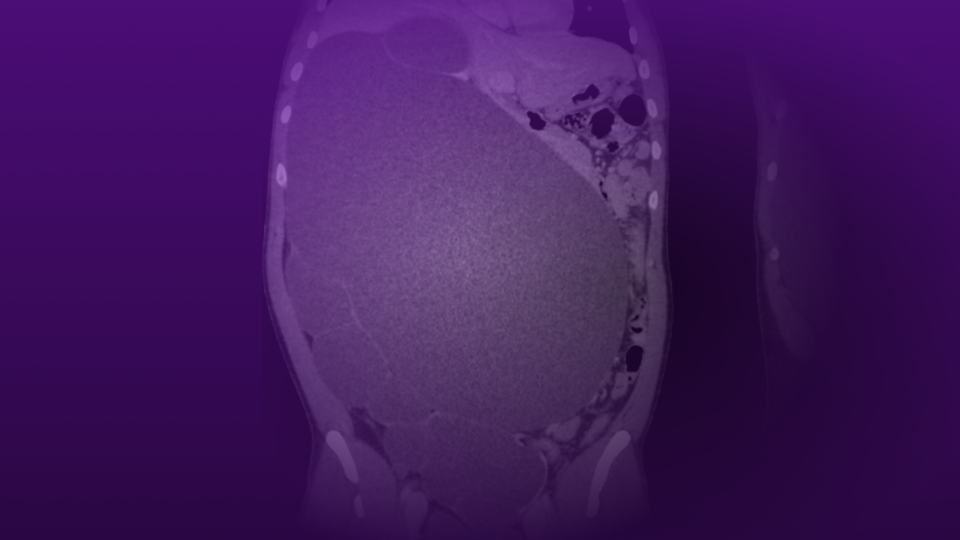
A recent computed tomography arterial portography (CTAP) scan showed a 1.5 cm right ureterovesical junction (UVJ) stone and 1.8 cm ureteropelvic junction (UPJ) stone with associated mild hydroureteronephrosis (Figure 1). Over 5 cm total stone burden was documented in each kidney, with stones in every calyx versus nephrocalcinosis (Figure 2).
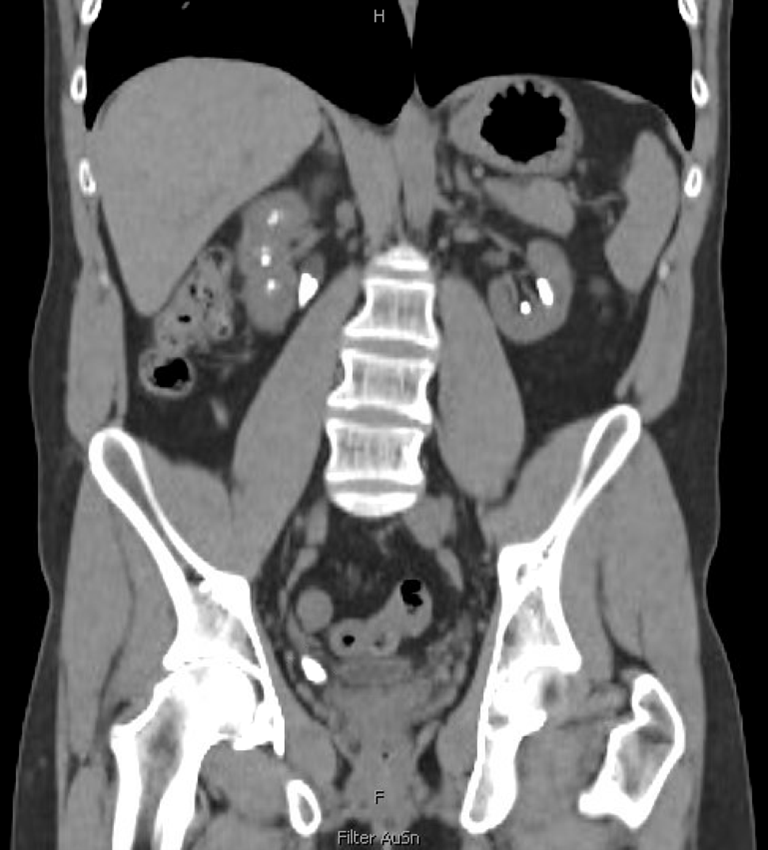
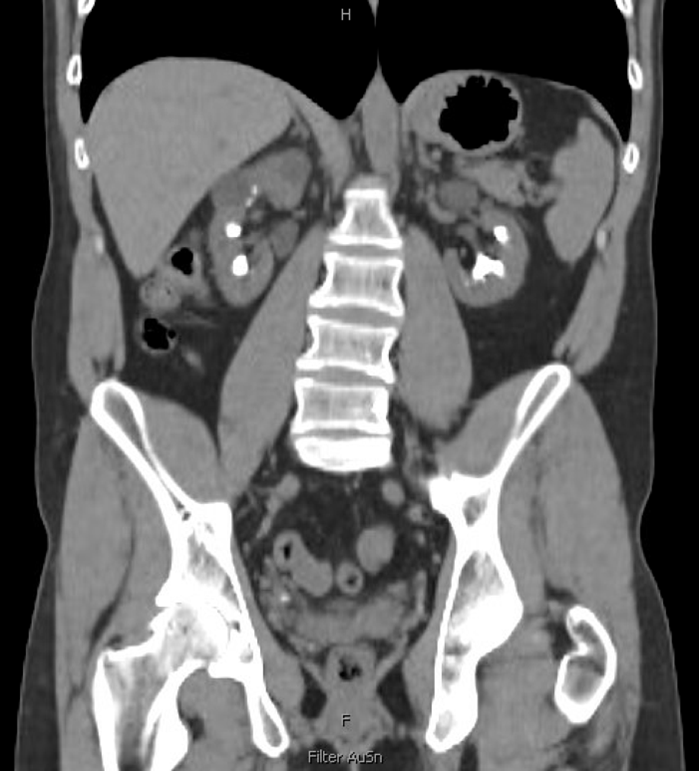
His baseline creatinine level was 4.37 mg/dL with a glomerular filtration rate (GFR) of 17.7 mL/min/1.73m2, indicating stage 4 CKD. His urinalysis showed large protein without blood, nitrite, or leukocyte esterase. He did not have any leukocytosis on bloodwork and denied any fevers or signs of infection.
Aside from two prior ureteroscopies, he did not have other medical or surgical history.
Shared Decision-Making
CTAP scans were reviewed and surgical treatment options were discussed. Although he was asymptomatic without renal colic, given the right renal obstruction from both UVJ and UPJ stones and already severely depressed renal function, surveillance was not an option. It was explained that prolonged renal obstruction can worsen kidney function and lead to further renal deterioration.
URS and PCNL details and surgical risks were discussed. He elected to undergo complete stone clearance with URS and laser lithotripsy of right UVJ stone as well as PCNL for right UPJ and right renal stones, followed a few weeks later by left stone clearance with PCNL.
Given the natural history of DD and eventual progression to complete renal failure and need for renal transplantation, the patient was made aware stone clearance will not prevent worsening renal function but may delay his need for transplant by a considerable interval. Understanding these risks and post-operative course, he elected to proceed with surgical removal of his stone burden.
Surgical Approach
He underwent uncomplicated right URS with lithotripsy and removal of UVJ stone (Figure 3). An open-ended catheter was placed to allow contrast roadmap of the right upper tract for percutaneous access. Although ultrasound guidance is becoming a mainstay for access placement, having retrograde pyelogram at time of percutaneous access allows for differentiation between nephrolithiasis and nephrocalcinosis and optimizes access tract to true intra-collecting system urolithiasis as opposed to intraparenchymal calcification.
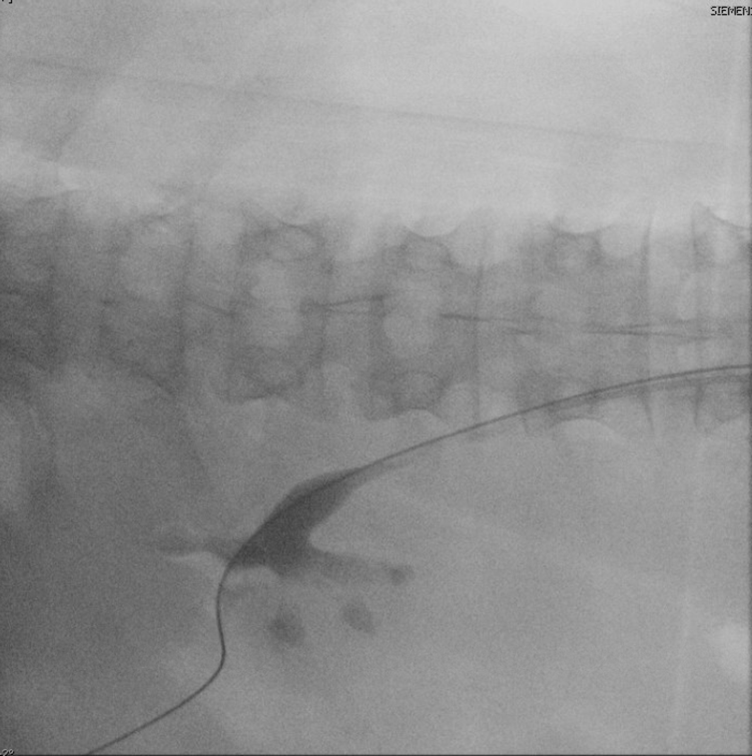
Access was obtained into the posterior upper pole with biplanar fluoroscopy and bull’s eye technique (Figure 4). A 24F access sheath was placed and complete right kidney stone clearance was achieved with both rigid and flexible nephroscopy utilizing a lithotriptor with ballistic and ultrasonic technology with built-in suctioning.
Direct vision and fluoroscopy showed complete stone-free status at end of PCNL (Figure 5). A ureteral stent was placed in the right ureter/kidney to maximize drainage and a temporary 18F nephrostomy tube was placed in the access tract.
The patient’s post-operative course was unremarkable and his nephrostomy tube was removed the following day prior to discharge. He followed up in the office five days later and underwent cystoscopy and right ureteral stent removal without issues. He reported minimal pain at incision site and resumed his normal activities one week out from his operation. His kidney function remained stable and his hematuria resolved a few days after stent removal.
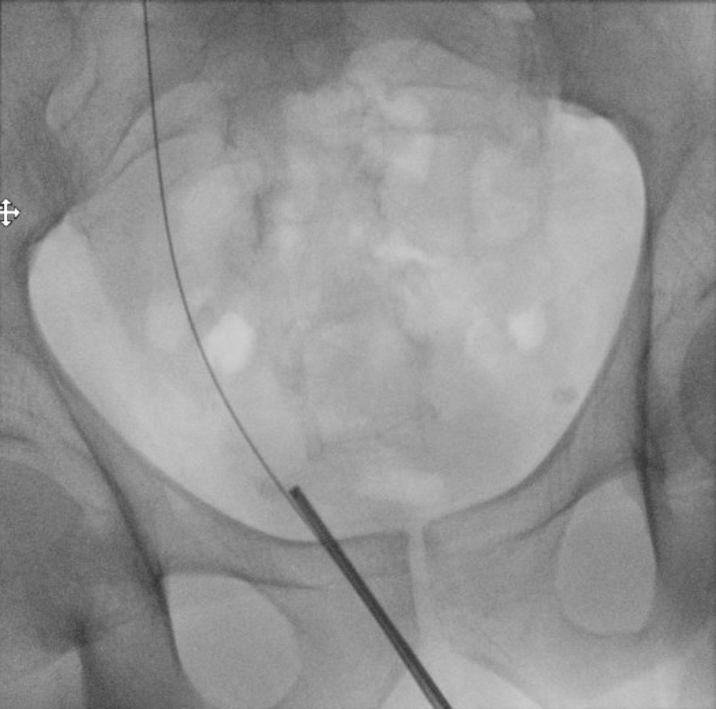
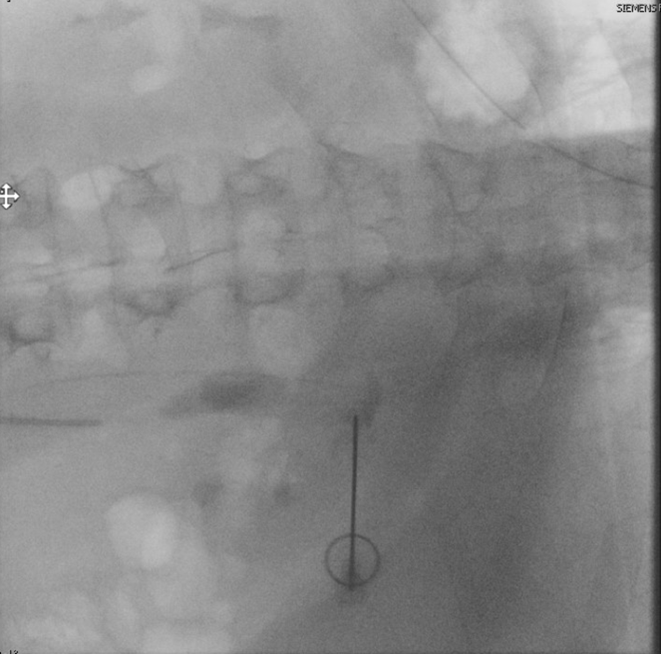
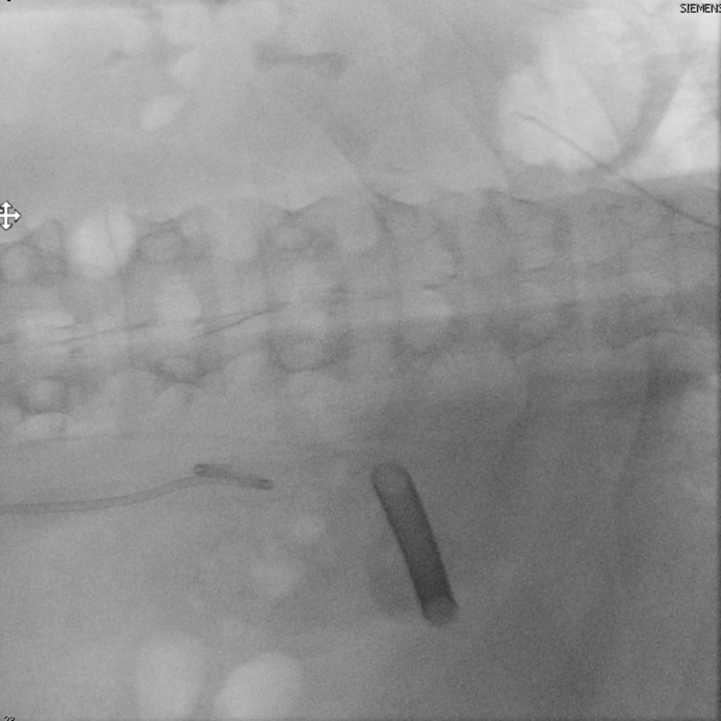
He returned three weeks later for stone clearance on the left side. Again, percutaneous access was obtained into the posterior upper pole using fluoroscopic guidance (Figure 6). A 24F ClearPetra (Karl Storz, Germany) suctioning nephrostomy sheath was placed and PCNL was performed.
There were more stones in calyces inaccessible by rigid nephroscope and stone clearance required use of flexible nephroscopy with laser lithotripsy. Having access with the tip-steerable ClearPetra sheath allowed for decreased stone scattering and antegrade migration as well as reduced intrarenal pressure. The EUA Guidelines advocate for suctioning during PCNL to decrease intrarenal pressure and increase stone-free rates4.
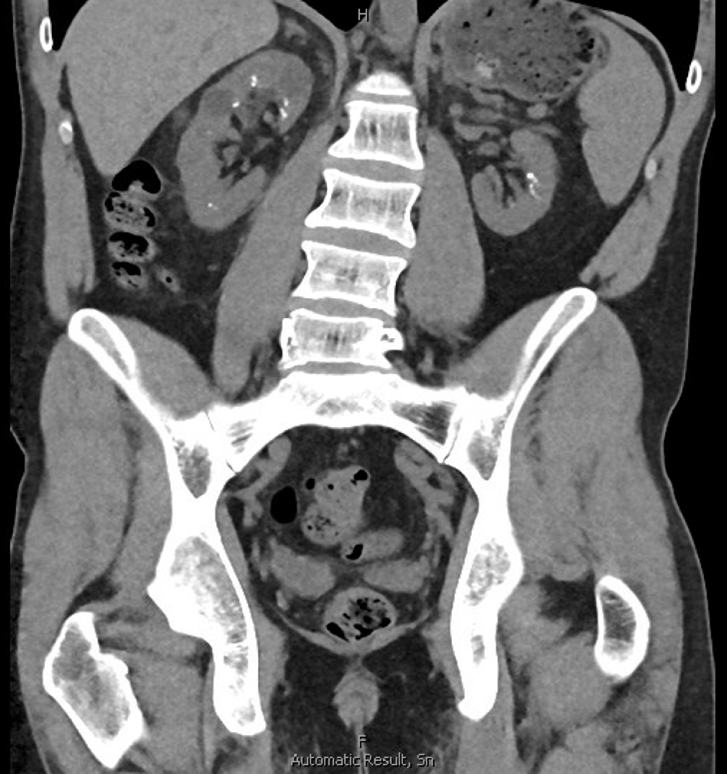
The patient underwent successful left PCNL and postoperative CTAP confirmed complete stone clearance on both sides (Figure 7). His creatinine level did uptrend immediately postop but follow-up a few weeks after surgery showed stable renal function with return to pre-operative GFR over 15 mL/min/1.73m2.
Stones were primarily calcium phosphate and he tolerated both operations well and returned to normal physical activities and sports after six days of recovery.
Conclusion
The patient presented in this case study had right ureteral stones with obstruction in addition to bilateral renal stone burden more than 5 cm on each side. We performed staged PCNL three weeks apart with removal of right ureteral stone with URS at time of initial PCNL and achieved complete stone clearance in both kidneys within one month, confirmed on post-operative CT scan.
Using one percutaneous access tract in each kidney and a combination of rigid and flexible nephroscopy and suction access sheath, all intra-collecting system renal stones were lithotripsied and removed. The tip-steerable suctioning nephrostomy sheath helped in reducing intrarenal pressure and immediate post-operative pain while increasing stone-free rate due to its Y-type connector with vacuum aspiration.
The patient had a successful outcome contributed by the application of new surgical technologies and multidisciplinary care at a comprehensive tertiary health care center. The patient will require close follow-up at frequent intervals with collaboration between nephrology and urology for management of this rare disease.
Discussion
Although the prevalence of DD is rare (1 in 400,000 to 1,000,000), its clinical manifestation can be devastating with 30 to 50 percent of affected males developing end-stage renal disease (ESRD) leading to dialysis5.
Diagnosis: DD is characterized and diagnosed by findings of low-molecular-weight (LMW) proteinuria, hypercalciuria, and at least one presentation of nephrolithiasis, nephrocalcinosis, hematuria, hypophosphatemia, or CKD6. Proteinuria is at least five times the upper limit of normal and usually more than 10 times in patients with DD. Hypercalciuria exhibits more than 4 mg calcium (0.1 mmol)/kg in 24 hours urine collection or more than 0.25 calcium/creatinine mg/mg (0.57 mmol/mmol) on spot urine test. Family history must also be consistent with X-linked inheritance. Differential diagnosis includes other proximal tubular dysfunctions, namely Fanconi syndrome, glomerulosclerosis, and Donnai-Barrow syndrome. Although renal biopsy can be performed as part of workup, biopsy alone is not definitive and is not required to make a diagnosis. Genetic testing is indicated in all patients with suspicion of DD to confirm diagnosis.
Dent Disease 1 (DD1) – CLCN5: About 60 percent of patients with DD have pathogenic variants on the CLCN5 gene, which encodes a family of voltage-gated chloride channels. These channels play a role in the control of membrane excitability, transepithelial transport, and cell volume expansion, but exact mechanisms are unknown7. DD1 causes disorder of the proximal renal tubules resulting in diffuse renal calcifications.
Dent Disease 2 (DD2) – OCRL: DD2 (15 percent of patients) involves pathogenic variants in the OCRL gene, which also causes the oculocerebrorenal syndrome of Lowe disease. The OCRL gene encodes an inositol polyphosphate 5-phosphatase, an enzyme which helps regulate membrane trafficking, actin polymerization, and promote cellular homeostasis. Half of patients with OCRL deficiency will have dense congenital cataracts and infantile glaucoma. There are other extrarenal manifestations of DD2; most affected males will have some degree of intellectual disability, short stature, proximal tubular dysfunction of Fanconi type, and glomerulosclerosis that progressively leads to CKD and ESRD by age 20. Over 42 pathogenic variants have been identified in males with DD2. The exact mechanism behind the transition from proximal tubular dysfunction to CKD/ESRD in DD2 remains unknown. It is postulated that early changes in proximal tubular cells—including proliferation, dedifferentiation, and autophagy—may become maladaptive and promote inflammation and progression of tubulointerstitial fibrosis8. Tubular proteinuria may also elicit stress responses in the distal nephrons.
Medical Management: No guidelines exist for treatment of DD, but the mainstays of therapy are to decrease hypercalciuria, prevent urolithiasis/nephrocalcinosis, and delay progression of CKD. Assessment of the extent of DD should be made by: 1) measuring renal function, 2) characterizing stone and intraparenchymal calcification burden by imaging (usually noncontract CTAP), 3) assessing bone disease and stunted growth in children, 4) evaluating for intellectual disability, 5) examining eyes for cataracts and visual impairment, and 6) consulting with a genetic counselor.
Treatment interventions center around decreasing hypercalciuria to limit rate of stone and intraparenchymal calcification growth. Citrate supplementation has been shown to slow CKD development in Clcn5-knockout mice and reduce recurrent calcium oxalate stones in patients with significant hypercalciuria and hypocitraturia9. Although there have been no human trials assessing citrate efficacy in DD, about a quarter of patients with DD are prescribed potassium citrate to reduce metabolic acidosis and slow CKD progression.
Phosphate supplementation is also useful in patients with hypophosphatemia. Prolonged hypophosphatemia may cause bone demineralization resulting in rickets in children and osteomalacia in adults. The hypophosphatemia seen in DD is generally mild, so it can be corrected with increased dietary phosphate and oral supplementation (20 mg/kg/day). Vitamin D supplementation is contraindicated as calcitriol levels are high and there is potential to worsen hypercalciuria. Vitamin A can be repleted with supplementation in patients with evidence of ocular symptoms and deficiency as retinol-binding protein (which carries serum vitamin A) is lost in LMW proteinuria.
Children with significant growth failure and start of CKD, despite correction of metabolic acidosis, may benefit from taking growth hormone. Growth is compromised in 27 percent and 54 percent of young patients with DD1 and DD2, respectively10. Height charts should be used to plot velocity and confirm growth retardation prior to initiating growth hormone use, especially in patients with DD2.
Thiazide treatment has been used with some success in patients with DD to improve hypercalciuria. Hydrochlorothiazide in doses greater than 0.4 mg/kg/day have led to decreased urinary calcium excretion by more than 40 percent in young men with DD11. However, there are significant side effects associated with thiazide use including hypokalemia, low blood pressure due to volume depletion, dizziness, and muscle aches/cramping from electrolyte imbalances. Patients with DD2 are especially more susceptible to severe dehydration and acute kidney injury (AKI); the benefits and risks of medication must be explored individually. A recent randomized controlled trial published in the New England Journal of Medicine showed no significant benefits of hydrochlorothiazide on recurrence rate of calcium stones in patients without DD, even in high doses of 50 mg/day12. Despite this, more than a third of patients with DD are treated with thiazides at some point due to lack of a more effective therapeutic regimen.
ACE inhibitors and angiotensin receptor blockers (ARB) have been used with some success in children with nephrotic-range proteinuria to prevent or delay further loss of renal function. However, their effectiveness is not well-established and they are not routinely prescribed for DD. No studies evaluating ACE inhibitors or ARBs on CKD progression has been studied in real patients or animal models. Although treatment with ACE inhibitors or ARBs may be beneficial for individuals with focal glomerulosclerosis, they are not thought to be helpful for the focal glomerulosclerosis that is associated with DD given the tubular nature of proteinuria rather than glomerular. Several small case series failed to demonstrate significant decrease in proteinuria during ACE inhibition and ARB treatment3.
Surgical Management: Surgical management of patients with significant nephrolithiasis follows the same principle for those without DD. Given the higher stone burden in patients with DD, URS and PCNL are both considered standard treatment options.
Renal Transplantation: There is no cure for DD and treatment ultimately culminates with renal transplantation for the majority of affected patients. Male family members of the patient are generally considered safe living donors, while mothers and half of sisters are carriers of the pathogenic variant and unsuitable as potential donors. Disease does not recur after successful transplant and most patients find a suitable donor (cadaveric or living) prior to requiring dialysis.
References
- Dent CE, Friedman M. Arch Dis Child. 1964;39(205):240-9. DOI.
- Hoopes RR Jr, et al. Am J Hum Genet. 2005;76(2):260-267. DOI.
- Blanchard A, et al. Kidney Int. 2016;90(2):430-439. DOI.
- Skolarikos A, et al. Eur Urol. 2025;88(1):64-75. DOI.
- Wrong OM, Norden AG, Feest TG. QJM. 1994;87(8):473-493. DOI.
- Bökenkamp A, et al. Nephrol Dial Transplant. 2025;40(5):852-864. DOI.
- Jentsch TJ, et al. Pflugers Arch. 1999;437(6):783-795. DOI.
- Liu BC, et al. Kidney Int. 2018;93(3):568-579. DOI.
- Cebotaru V, et al. Kidney Int. 2005;68(2):642-652. DOI.
- Gianesello L, et al. Hum Genet. 2021;140(3):401-421. DOI.
- Raja KA, et al. J Am Soc Nephrol. 2002;13(12):2938-2944. DOI.
- Dhayat NA, et al. N Engl J Med. 2023;388(9):781-791. DOI.