
Resolving a Complex Cascade of Symptoms
A 35-year-old female was admitted via the ED at NYU Langone Health’s Tisch Hospital with one week of fever, nausea, intense abdominal pain, and diffuse bone pain. She had a history of systemic lupus erythematosus (SLE), secondary antiphospholipid syndrome (APS) with a triple-positive aPL antibody profile, HELLP syndrome, and chronic thrombocytopenia.
Given the patient’s medical history, lab and imaging results, fever, and intense pain, H. Michael Belmont, MD, co-director of the Lupus Center, concluded that her presentation reflected a flare of atypical catastrophic APS (CAPS) with widespread thrombosis-related ischemia with involvement in her bone, bone marrow, and hepatobiliary tract.
“This patient was highly complex, with her lupus and secondary antiphspholipid syndrome unmasking an underlying genetic defect that also gave her complement-mediated HUS.”
H. Michael Belmont, MD
Worsening symptoms, including acute bilateral blurry vision and a nonoliguric acute kidney injury with nephrotic range proteinuria, led Dr. Belmont and colleagues to order atypical hemolytic uremic syndrome (aHUS) genetic testing. The test revealed variants in both the CFB and MCP (CD46) genes for complement regulatory proteins.
“This patient was highly complex, with her lupus and secondary antiphospholipid syndrome unmasking an underlying genetic defect that also gave her complement-mediated HUS,” he says.
Effectively managing and eventually resolving the case relied on unveiling and treating the atypical HUS and consulting with specialists in multiple departments to address the refractory CAPS complicated by bone, bone marrow, liver, biliary, choroid/retina, kidney, and jejunum involvement.
One month after her hospital admission, the patient was well enough to be discharged. “Close and frequent consultations allowed our interdisciplinary care team to quickly respond to an evolving and cascading suite of symptoms,” says Dr. Belmont.
A Highly Complicated Presentation
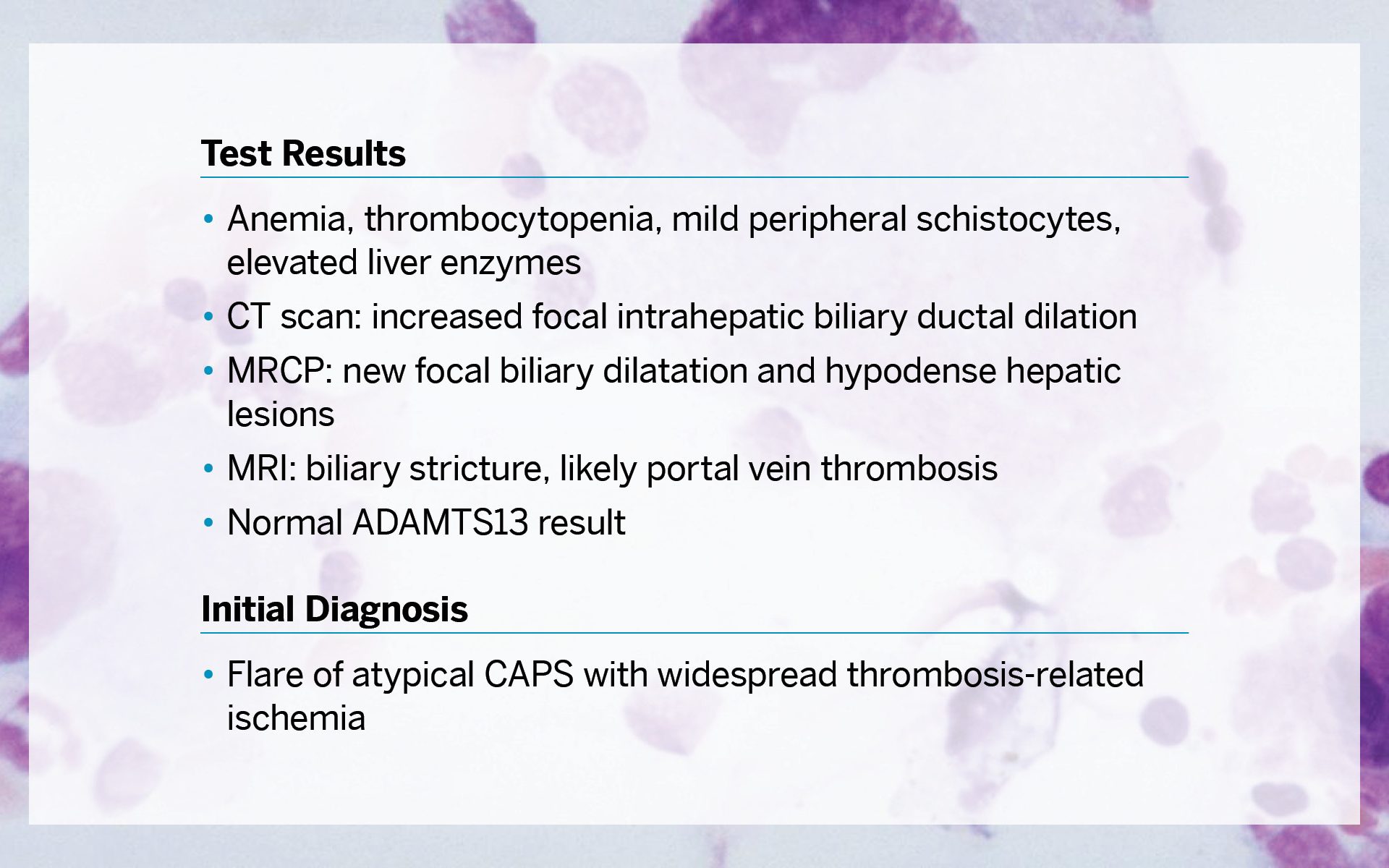
The patient’s test results revealed a highly complicated presentation and included anemia, thrombocytopenia, mild peripheral schistocytes, and elevated liver enzymes. A CT scan showed increased focal intrahepatic biliary ductal dilation; magnetic resonance cholangiopancreatography (MRCP) showed new focal biliary dilation and hypodense hepatic lesions; and MRI showed biliary stricture, likely portal vein thrombosis. The ADAMTS13 result was normal. The initial diagnosis was flare of atypical catastrophic antiphospholipid syndrome (CAPS) with widespread thrombosis-related ischemia.
Managing a Cascading Crisis
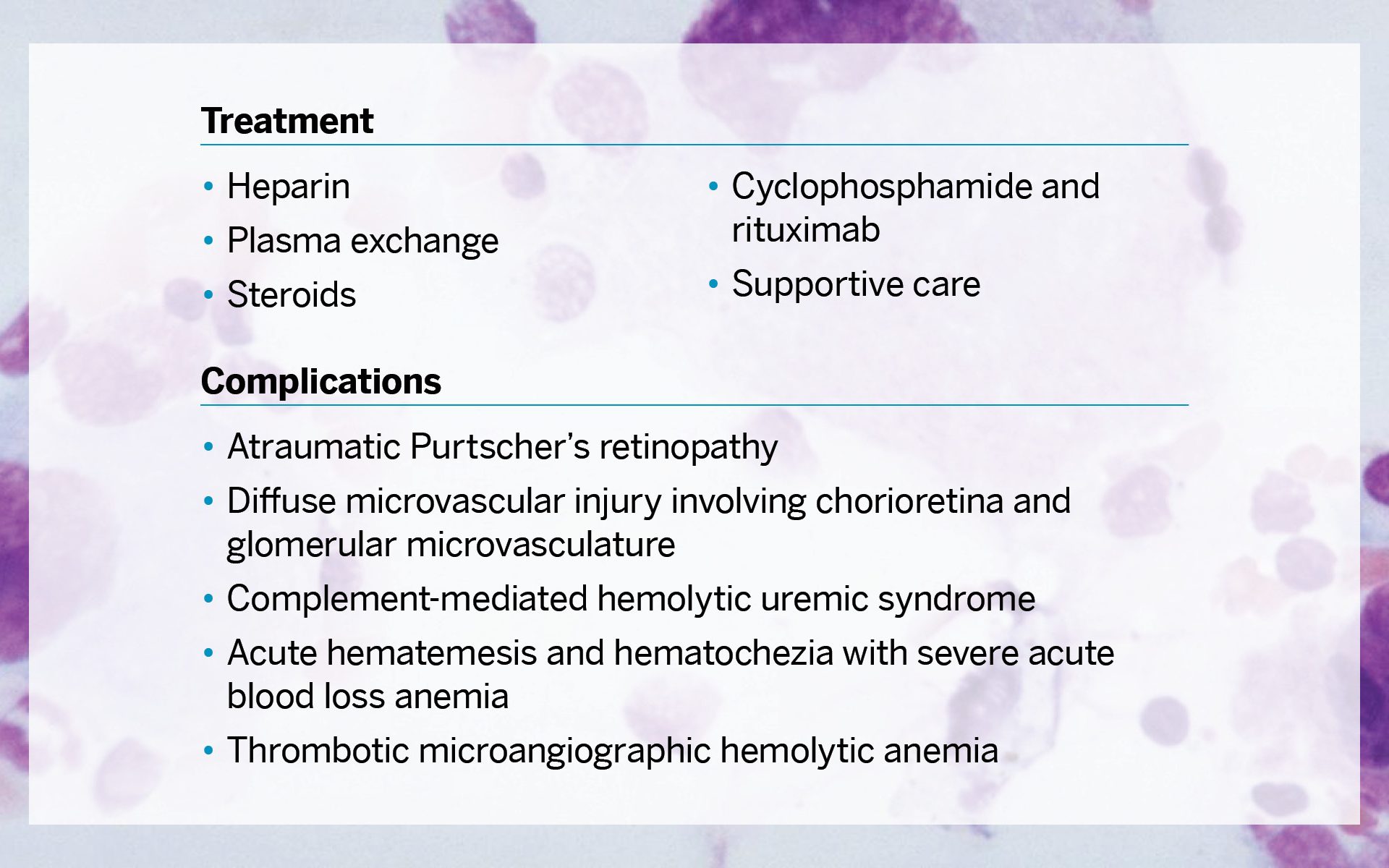
To manage a cascading crisis, treatment included heparin, plasma exchange, steroids, cyclophosphamide and rituximab, and supportive care. However, complications included atraumatic Purtscher’s retinopathy, diffuse microvascular injury involving chorioretina and glomerular microvasculature, complement-mediated hemolytic uremic syndrome, acute hematemesis and hematochezia with severe acute blood loss anemia, and thrombotic microangiographic hemolytic anemia.
Interdisciplinary Treatment
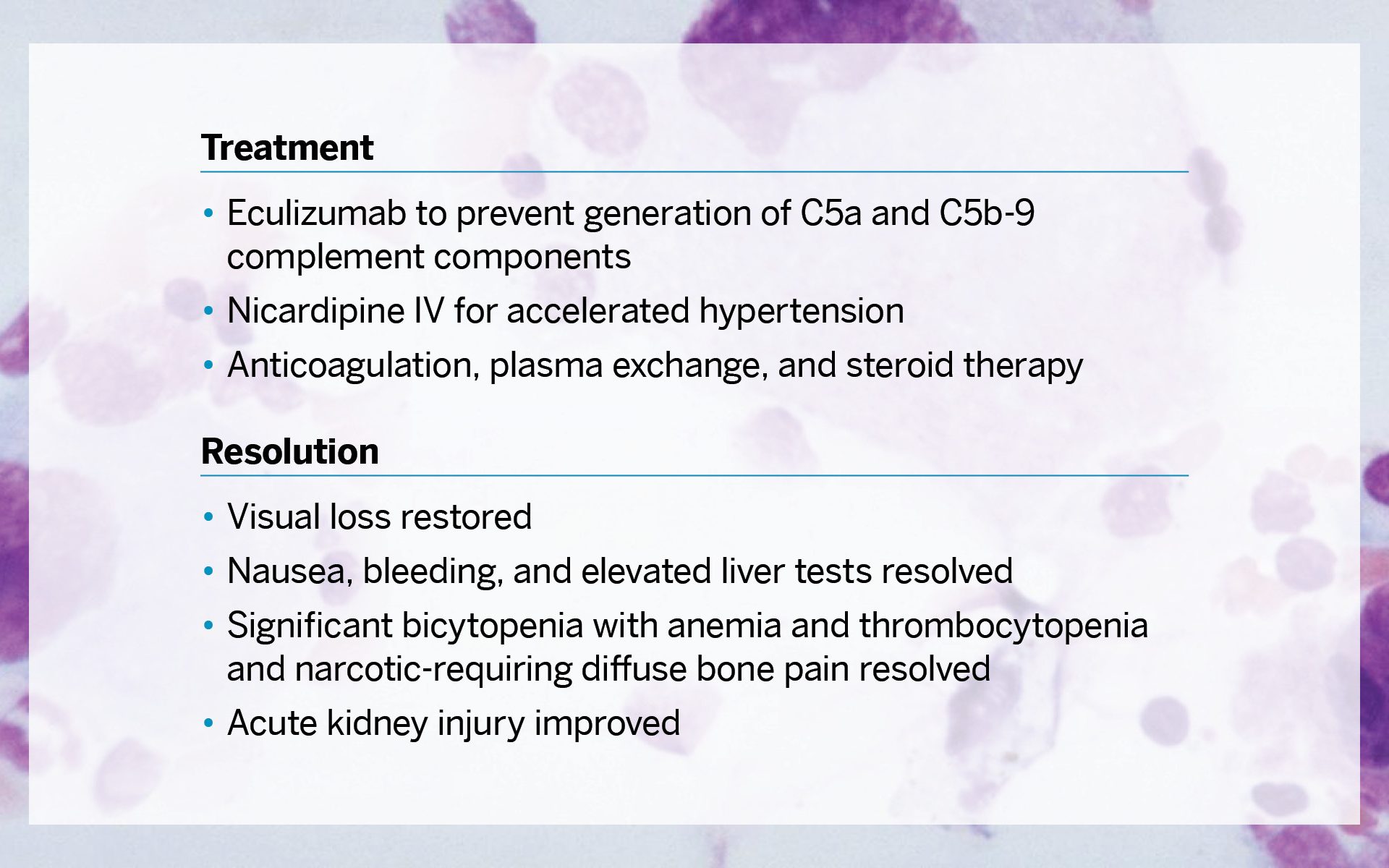
Interdisciplinary treatment was required and included eculizumab to prevent generation of C5a and C5b-9 complement components, nicardipine IV for accelerated hypertension, and anticoagulation, plasma exchange, and steroid therapy. For the resolution, the patient’s visual loss was restored; nausea, bleeding, and elevated liver tests were resolved; significant bicytopenia with anemia and thrombocytopenia and narcotic-requiring diffuse bone pain were resolved; and the acute kidney injury improved.



